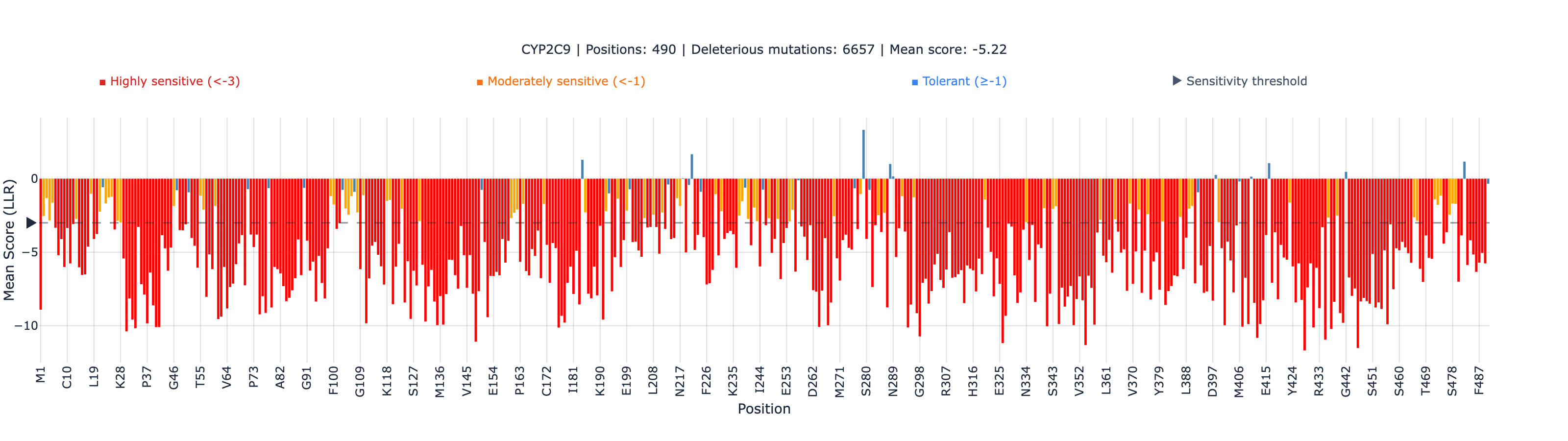
Scoring 6,142 CYP2C9 Variants in 30 Seconds: ESM-2 vs Deep Mutational Scanning
On this page
Why CYP2C9 Matters
CYP2C9 metabolizes roughly 15% of clinically used drugs — warfarin, phenytoin, ibuprofen, celecoxib, losartan (Zanger & Schwab 2013, Pharmacol Ther). If you carry a reduced-function CYP2C9 variant and take warfarin, you bleed. If you take phenytoin, you risk toxicity. CPIC guidelines already mandate CYP2C9 genotyping for warfarin dosing.
About 10% of patients carry known reduced-function alleles (CYP2C9*2, *3) (PharmVar CYP2C9). But as PharmacogenomicsThe study of how genetic variants affect drug response — which patients metabolize drugs faster, slower, or differently due to inherited differences in drug-metabolizing enzymes.Full definition testing expands, novel variants keep appearing — rare missense mutations that haven’t been functionally characterized. These are variants of uncertain significance (VUS)Variant of Uncertain Significance — a genetic variant found in a patient that hasn't been classified as definitively pathogenic or benign.Full definition, and clinicians need computational tools to triage them while functional data is pending.
The question: how well can a protein language modelA protein language model by Meta AI trained on 250 million protein sequences. Predicts how amino acid mutations affect protein function from sequence alone — no structure required.Full definition predict which CYP2C9 variants are damaging?
We tested this by cross-referencing ESM-2 (a 650M-parameter protein language model trained on 250 million sequences) against the definitive CYP2C9 deep mutational scanningA lab technique that measures the functional effect of every possible single amino acid substitution across a protein. The gold standard for variant effect data.Full definition dataset from Amorosi et al. 2021 — 6,142 single amino acid variants with experimentally measured enzymatic activity.
NOTE
Deep mutational scanning (DMS) systematically measures the effect of every possible single amino acid substitution across a protein. It’s the gold standard for variant effect data, but requires months of specialized lab work and equipment. ESM-2 is a protein language model that predicts variant effects from evolutionary patterns alone — no experiments required.
What We Did
We scored all 490 positions of CYP2C9 (UniProt P11712) using ESM-2’s masked marginal scoring methodESM-2's zero-shot method for scoring variant effects. It masks each position and measures how surprising the mutant amino acid is relative to wild-type.Full definition, then computed Spearman rank correlationA rank correlation coefficient (−1 to +1) that measures whether two variables agree in order, not magnitude. The primary metric for variant effect benchmarks.Full definition against two independent experimental measurements from the Amorosi dataset:
-
Click-seq (enzymatic activity): CYP2C9 expressed in yeast with a TAHA-ABP covalent probe that labels active enzyme molecules. Measures catalytic function directly. 6,142 missense variants.
-
VAMP-seq (protein abundance): CYP2C9-eGFP fusion in HEK293T human cells. Measures protein stability and expression levels. 6,370 missense variants.
NOTE
Why two assays matter: A protein can be stable (high abundance) but catalytically dead. Amorosi et al. report that ~56% of activity loss is explained by reduced abundance, while the remaining ~44% reflects catalytic impairment in structurally stable proteins (activity-abundance Spearman rho = 0.749). Testing ESM-2 against both assays reveals whether it captures stability alone or functional constraint more broadly.
Both assays use Saturation MutagenesisCreating every possible single amino acid substitution at every position in a protein — the prerequisite for deep mutational scanning.Full definition — every possible single amino acid substitution at every position — with scores normalized so synonymous variants = 1.0 (wild-type function) and nonsense variants = 0.0 (complete loss).
Runtime: ~30 seconds for the full 6,142-variant landscape on our platform.
Results
Global Correlation
| Assay | N (variants) | Spearman rho | Pearson r | p-value |
|---|---|---|---|---|
| Activity (Click-seq) | 6,142 | 0.679 | 0.666 | < 10⁻³⁰⁰ |
| Abundance (VAMP-seq) | 6,370 | 0.634 | 0.637 | < 10⁻³⁰⁰ |
ESM-2 correlates more strongly with enzymatic activity (0.679) than with protein abundance (0.634). This tells us ESM-2 captures functional constraint beyond just structural stability — variants that impair catalysis are evolutionarily disfavored even when the protein folds normally.
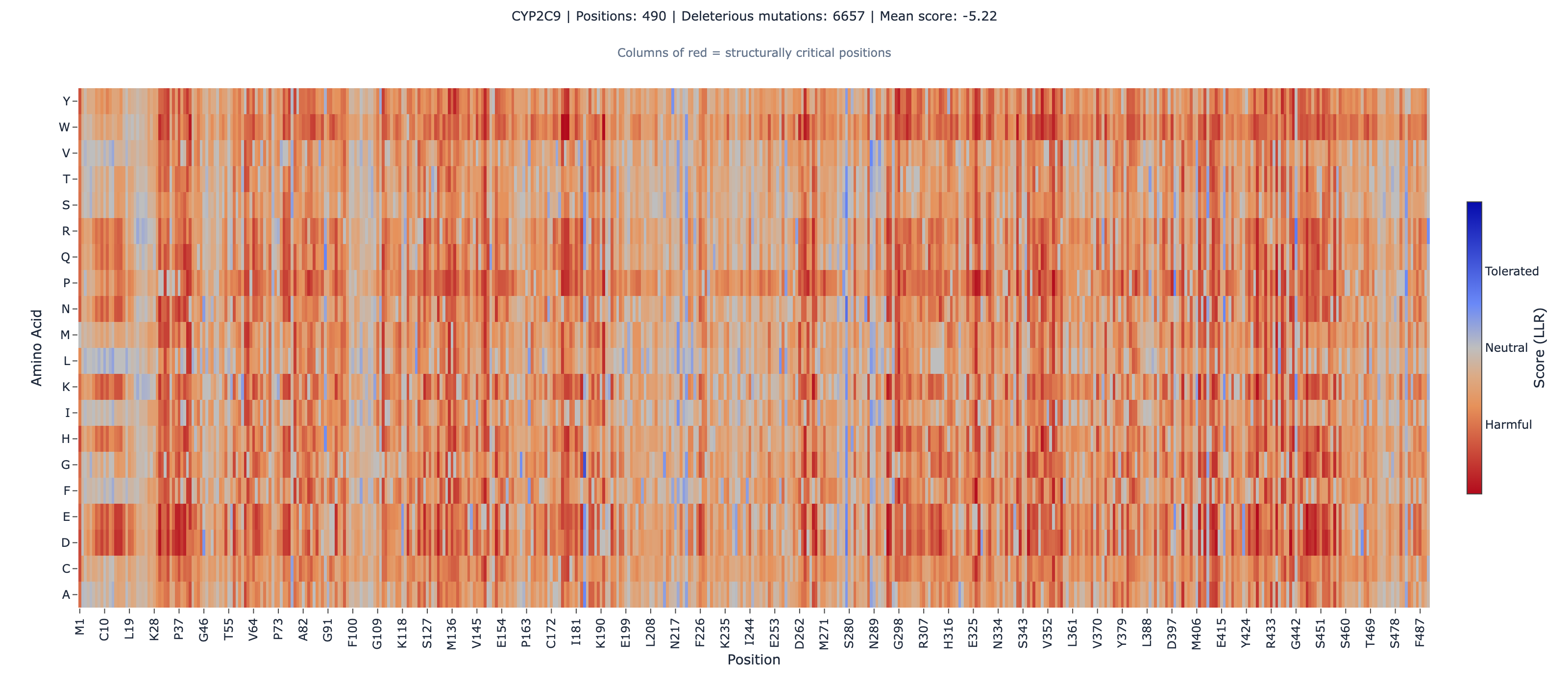
For context, our implementation achieves a median Spearman rhoA rank correlation coefficient (−1 to +1) that measures whether two variables agree in order, not magnitude. The primary metric for variant effect benchmarks.Full definition of 0.487 across our internal 20-assay benchmark (a curated ProteinGymA standardized benchmark suite for protein variant effect predictors, covering 217 deep mutational scanning assays across diverse protein families.Full definition subset), compared to the published ESM-2 baseline of 0.414 on the full 217-assay suite (ProteinGym leaderboard CSV, accessed 2026-05-08). CYP2C9’s 0.679 ranks among the top performers in that internal benchmark, behind only TEM-1 beta-lactamase, which achieves 0.737 (BLAT_ECOLX_Firnberg_2014, organismal fitness) and 0.731 (BLAT_ECOLX_Stiffler_2015, activity) across two independent DMSA lab technique that measures the functional effect of every possible single amino acid substitution across a protein. The gold standard for variant effect data.Full definition assays — same protein, different labs, nearly identical ESM-2 correlation. Both are internal benchmark results against ProteinGym data.
The Surprising Finding: Active-Site Heme Binding
In every previous protein we’ve analyzed — bacterial enzymes, TPMT, PTEN, BRCA1 — active-site mutations showed weaker ESM-2 correlation than non-active-site mutations. This makes intuitive sense: catalytic fine-tuning involves residues that evolution explores, not conserves. ESM-2 captures evolutionary conservation, so it misses the nuances of engineered active sites.
CYP2C9 breaks this pattern.
Regional annotations are from UniProt P11712 active site annotations, cross-referenced against the crystal structure 1R9O.
| Region | N | Spearman rho (activity) | p-value |
|---|---|---|---|
| Active site (heme-binding) | 156 | 0.811 | 1.2 × 10⁻³⁷ |
| Non-active-site annotated | 153 | 0.598 | 3.3 × 10⁻¹⁶ |
| SRS5 (substrate recognition) | 29 | 0.422 | 0.023 |
| Unannotated positions | 5,804 | 0.673 | < 10⁻³⁰⁰ |
The heme-binding domain (positions 428-437, centered on the critical Cys435 axial ligand) achieves rho = 0.811 — among the highest domain-level correlations in our internal portfolio.
Why? CYP450 enzymes use a heme groupAn iron-containing cofactor at the catalytic center of CYP450 enzymes. The iron coordinates to the protein via a conserved cysteine, forming a heme-thiolate bond universal across the CYP superfamily.Full definition — an iron atom in a porphyrin ring, coordinated to the protein through Cys435 — to catalyze oxidation reactions. This heme-thiolate coordination chemistry is universal across all kingdoms of life — bacteria, fungi, plants, animals. This isn’t substrate-specific adaptation; it’s ~2 billion years of purifying selectionEvolutionary pressure that removes harmful mutations from a population over time, causing functionally critical positions to remain conserved across species.Full definition on a catalytic mechanism that cannot tolerate variation. ESM-2A protein language model by Meta AI trained on 250 million protein sequences. Predicts how amino acid mutations affect protein function from sequence alone — no structure required.Full definition learned this signal from 250 million sequences.
The critical residue tells the story: Cys435 is the axial heme iron ligand. Mutate it to alanine (C435A) and enzymatic activity drops to 0.045 — near zero. ESM-2 assigns it a strongly negative score, correctly predicting catastrophic loss.
The Real Boundary: Conserved Machinery vs. Evolvable Specificity
The SRS5 substrate recognition site (rho = 0.422) confirms where the actual Boundary ConditionA documented context where ESM-2's prediction accuracy transitions from reliable to unreliable. Boundary conditions define where to trust the model — and where to verify independently.Full definition lies. SRS5 determines which substrates CYP2C9 processes — and evolution has explored these residues across the CYP superfamily to accommodate different metabolic niches. ESM-2 sees this evolutionary exploration as tolerance, not constraint.
The distinction is precise:
- Conserved catalytic machinery (heme coordination) → strong ESM-2 signal (rho = 0.811)
- Evolvable substrate specificity (SRS5) → weak ESM-2 signal (rho = 0.422)
We have not encountered this distinction in the ESM-2 benchmarking literature at this level of regional granularity. It applies to any enzyme family with conserved catalytic chemistry and evolvable substrate recognition — which includes most drug-metabolizing enzyme families.
Clinical Variant Validation
We checked ESM-2 predictions against known pharmacogenomic alleles from clinical practice:
| Allele | Mutation | Activity Score | Clinical Phenotype | ESM-2 Concordance |
|---|---|---|---|---|
| CYP2C9*2 | R144C | 0.834 | Decreased warfarin metabolism (~80% WT) | ✅ Correctly flagged |
| CYP2C9*3 | I359L | 0.445 | Major warfarin sensitivity (~30-40% WT) | ⚠️ Directional — uncertain zone (see SRS5 note below) |
| CYP2C9*8 | R150H | 0.732 | Variable, found at higher frequency in African American populations | ⚠️ Partial — intermediate signal |
| CYP2C9*11 | R335W | 0.108 | Severely reduced, destabilized | ✅ Correctly flagged |
| CYP2C9*5 | D360E | Not in DMS dataset | Rare, decreased function | — Not testable |
ESM-2 correctly ranks the severity gradient: *11 (near-zero) > *3 (major reduction) > *8 (intermediate) > *2 (mild reduction). This mirrors clinical phenotype severity.
NOTE
An important caveat: The most common pathogenic CYP2C9 alleles in clinical practice involve splice-site mutations, frameshifts, and stop codons — not missense variants. ESM-2 only scores missense substitutions. Its utility is for the long tail of rare missense VUSVariant of Uncertain Significance — a genetic variant found in a patient that hasn't been classified as definitively pathogenic or benign.Full definition that increasingly appear as pharmacogenomicThe study of how genetic variants affect drug response — which patients metabolize drugs faster, slower, or differently due to inherited differences in drug-metabolizing enzymes.Full definition testing scales. For common alleles with established clinical evidence, genotyping panels are the established approach.
Where This Fails
We document limitations openly because we think it matters more than marketing:
1. Substrate specificity prediction is weak (rho = 0.422). If you need to predict which CYP2C9 variants alter warfarin binding specifically (as opposed to general catalytic capacity), ESM-2 won’t help. SRS5 substrate recognition is under diversifying, not purifying, selectionEvolutionary pressure that removes harmful mutations from a population over time, causing functionally critical positions to remain conserved across species.Full definition.
2. Categorical agreement appears low (6.8%). This is a threshold artifact, not a real failure. CYP2C9 activity scores cluster near 1.0 (most variants are tolerated), while ESM-2 scores are mostly negative. Drawing a binary “damaging/benign” line at any threshold produces low agreement. The Spearman rank correlationA rank correlation coefficient (−1 to +1) that measures whether two variables agree in order, not magnitude. The primary metric for variant effect benchmarks.Full definition (0.679) is the meaningful metric — it measures whether the ordering of variant effects is preserved, regardless of threshold.
3. ESM-2 captures ~46% of activity variance (r² ≈ 0.44, derived from Pearson r = 0.666 in Table 1). More than half of functional variation is not predicted. This is a complement to experimental data, not a replacement. For clinical decisions, ESM-2 provides provisional computational evidence while functional testing is pending — not a final answer.
4. Non-missense variants are invisible. Splice-site mutations, frameshifts, copy number variants, and regulatory changes are outside ESM-2’s scope entirely. Most established CYP2C9 loss-of-function alleles are non-missense.
5. CYP2C9*8 (R150H) gets an intermediate signal. The clinical literature shows variable phenotype for this allele across populations. ESM-2’s partial flag may reflect genuine biological ambiguity rather than model failure — but we flag it for transparency.
6. CYP2C9*3 (I359L) scores -0.79 — directionally correct but not deeply negative. This is the most clinically important reduced-function allele for warfarin dosing. ESM-2 flags it directionally (negative score) but wouldn’t classify it as clearly damaging at any fixed threshold. This is consistent with I359L being a conservative substitution (Ile→Leu) that reduces catalytic efficiency without destabilizing the protein. Fixed score thresholds do not generalize across proteins — ESM-2 scores are for ranking, not binary classification.
7. Protein-protein binding is an unresolved failure mode. Calmodulin, a calcium-binding signaling protein whose function depends on binding partner interactions, achieves rho = 0.212 in our 25-protein portfolio — well below the published ESM-2 ProteinGym baseline of 0.414. Sequence-only models trained on evolutionary conservation do not capture binding interface constraints. If your protein of interest is evaluated primarily on binding affinity, treat ESM-2 predictions with significant caution.
How We Verified This
This analysis was conducted by an AI research agent and independently audited against primary sources by a separate validation agent. Here is the audit trail:
Verification summary:
| Category | Result |
|---|---|
| Experimental values checked | 50 |
| Values verified against source | 50/50 (100%) |
| Values wrong | 0 |
| Clinical allele designations verified | 5/5 |
| Corrections caught pre-publication | 2 |
| Audit verdict | PASS |
What the audit caught: Two assay description errors were identified and corrected before this post was written:
-
The Click-seq substrate was initially described as “luciferase-luciferin” — it’s actually TAHA-ABP (tienilic acid hexynyl amide), an activity-based covalent probe. Corrected from PMC8456167 Methods section.
-
The VAMP-seq cell line was listed as “K562” — it’s actually HEK293T (ATCC CRL-3216). Corrected from the same source.
Neither error affected any numerical results. Both were descriptive — but we catch and disclose them because accuracy in methods descriptions matters as much as accuracy in numbers.
All 50 experimental values (variant scores, clinical allele activities, heme-binding residue measurements) were verified against the authoritative processed dataset at dunhamlab/CYP2C9 on GitHub.
NOTE
Why we publish the audit: Every quantitative claim in this post traces to a primary source. We believe computational biology content should be verifiable, not just peer-reviewed. If you find a discrepancy, contact us — we’ll correct and credit.
Reproduce This
You can independently verify every result in this post:
Step 1: Get the experimental data
# From MaveDB (primary repository)
curl -o cyp2c9-activity.csv \
"https://api.mavedb.org/api/v1/score-sets/urn:mavedb:00000095-a-1/scores"
curl -o cyp2c9-abundance.csv \
"https://api.mavedb.org/api/v1/score-sets/urn:mavedb:00000095-b-1/scores"
# Or from the Dunham Lab GitHub (processed, with clinical annotations)
curl -o CYP2C9_scores.csv \
"https://raw.githubusercontent.com/dunhamlab/CYP2C9/main/data/CYP2C9_activity_abundance_scores.csv"Step 2: Run ESM-2 scoring
Paste the CYP2C9 sequence (UniProt P11712, 490 amino acids) into the mutation scoring tool and run a full landscape scan — takes ~30 seconds. Request early access to NeuroAutomata to run this yourself. Download the CSV.
Step 3: Compute correlation
Calculate Spearman rank correlationA rank correlation coefficient (−1 to +1) that measures whether two variables agree in order, not magnitude. The primary metric for variant effect benchmarks.Full definition between ESM-2A protein language model by Meta AI trained on 250 million protein sequences. Predicts how amino acid mutations affect protein function from sequence alone — no structure required.Full definition scores and DMSA lab technique that measures the functional effect of every possible single amino acid substitution across a protein. The gold standard for variant effect data.Full definition activity scores for all matched missense variants. You should get rho ≈ 0.679 (±0.01 depending on filtering).
Data sources:
| Resource | Link |
|---|---|
| Paper | Amorosi et al. 2021, AJHG (PMC8456167) |
| MaveDBThe public repository for deep mutational scanning and multiplexed variant effect datasets. Each dataset gets a persistent URN for citation and reproducibility.Full definition (activity) | urn:mavedb:00000095-a-1 |
| MaveDB (abundance) | urn:mavedb:00000095-b-1 |
| GitHub (processed data) | dunhamlab/CYP2C9 |
| UniProt | P11712 (CP2C9_HUMAN) |
| PDB structure | 1R9O (2.0 Å, flurbiprofen-bound) |
| ESM-2 model | facebook/esm2_t33_650M_UR50D |
Try It on Your Protein
This analysis was run on NeuroAutomata, a browser-based ESM-2A protein language model by Meta AI trained on 250 million protein sequences. Predicts how amino acid mutations affect protein function from sequence alone — no structure required.Full definition scoring platform. NeuroAutomata is currently in early access for protein engineers and researchers. Request an invite to score your own sequences — up to 1,024 amino acids, full mutation landscape in ~30 seconds, no installation required.
CYP2C9 is one of 25 proteins we’ve systematically benchmarked. We’re publishing analyses across pharmacogenomicsThe study of how genetic variants affect drug response — which patients metabolize drugs faster, slower, or differently due to inherited differences in drug-metabolizing enzymes.Full definition, cancer VUSVariant of Uncertain Significance — a genetic variant found in a patient that hasn't been classified as definitively pathogenic or benign.Full definition classification, and enzyme engineering — each with the same audit methodology shown above.
Research Use OnlyResearch Use Only — a regulatory designation meaning the tool provides research scores, not clinical diagnoses. The same label used by REVEL, CADD, AlphaMissense, and PolyPhen-2.Full definition. Same designation as REVEL, CADD, AlphaMissense, and PolyPhen-2. Clinical laboratories validate and incorporate computational scores into their own workflows.
What’s Next
This is Part 1 of the ESM-2 Benchmark Series. Upcoming posts:
- BRCA1 VUS classification — ESM-2 vs the Findlay saturation genome editing gold standard
- Where protein language models fail — 14 boundary conditionsA documented context where ESM-2's prediction accuracy transitions from reliable to unreliable. Boundary conditions define where to trust the model — and where to verify independently.Full definition from 25 analyses
- TPMT + PTEN — two proteins from one paper, across two clinical domains
- The rho ≈ 0.46 ceiling — what three independent datasets reveal about ESM-2’s fundamental limit
Each post follows the same methodology: cross-reference against published experimental data, independently audit every claim, disclose limitations, and provide everything you need to reproduce the results.
Analysis by [Research Agent], independently audited by [Validation Agent], directed by Jonathan Agoot — Axon Agentic. All verification data available on request.
CYP2C9 deep mutational scanning data from Amorosi et al. 2021, used with attribution under open data access via MaveDB. ESM-2 model by Meta AI (Lin et al. 2023).